Untangling a sticky mystery: Researchers make precious headway into a genetic form of Alzheimer’s disease
UC Santa Barbara researchers and collaborators in Colombia, Brazil and Germany are progressing toward an understanding of mechanisms that underlie Alzheimer’s Disease, in particular an early-onset, genetic form that has afflicted generations of an extended family in Colombia. They also shed some light on a woman from that family who managed to beat the odds.
“What are the chances,” said UCSB neuroscientist Kenneth S. Kosik, a senior author of a paper that appears in the journal Neuron. “It’s unbelievable serendipity.”
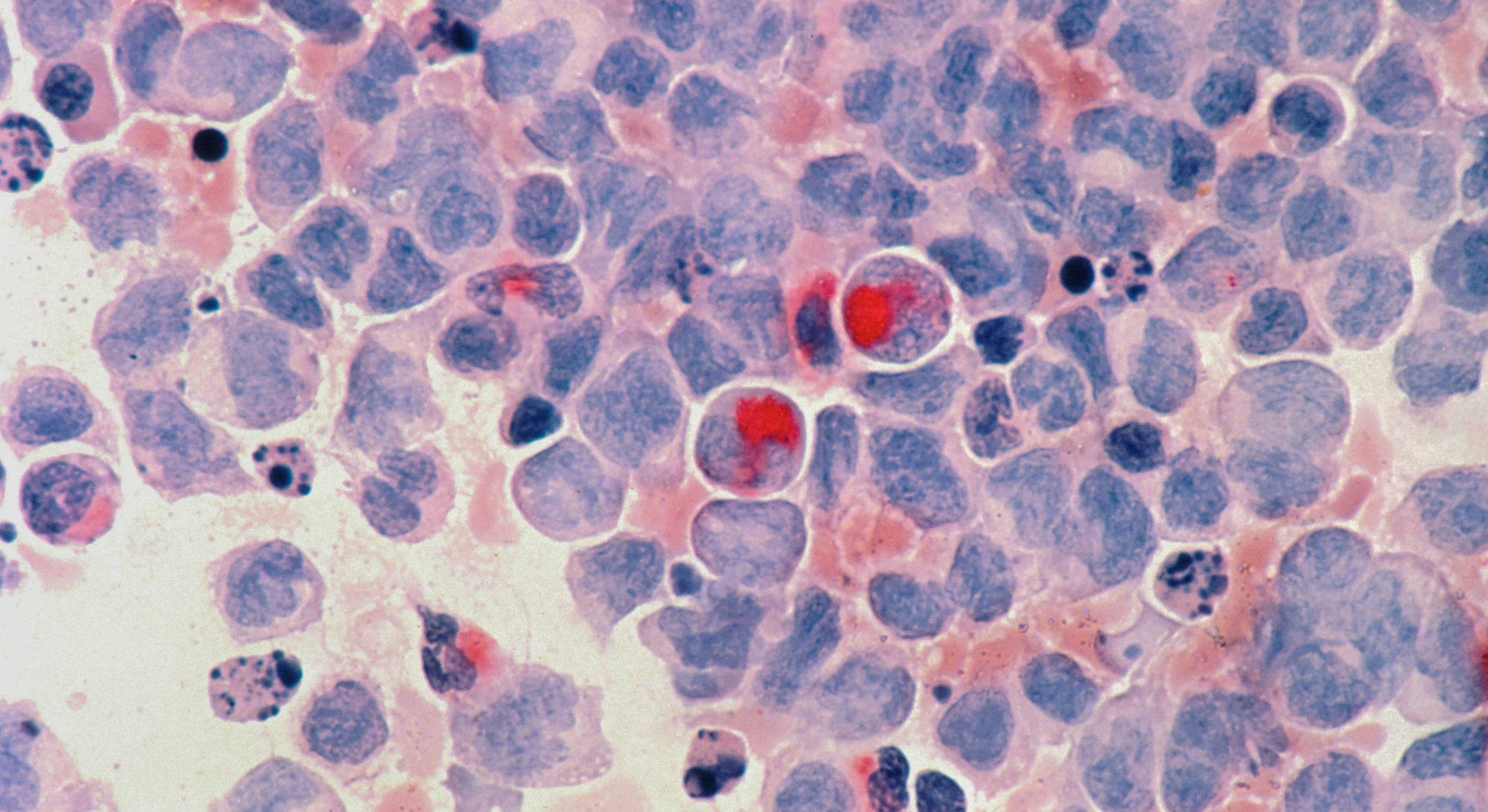
It all takes place in the rural mountain communities on the outskirts of Medellín in the department of Antioquia, where Kosik has been collaborating with Colombian neuroscientist Francisco Lopera for decades to study the family, members of which, like clockwork, begin to present the signs of Alzheimer’s dementia in their mid-40s. Genetic testing revealed that they each carry a mutation, called the paisa mutation, in their PSEN1 gene. The mutation, labeled E280A, is linked to the accelerated development of the sticky plaques between neurons that would otherwise likely develop late in life. These plaques, in addition to tangles of a misfolded structural protein inside neurons called tau, are the hallmarks of Alzheimer’s disease.
What makes this family important, aside from their heroic willingness to work jointly with researchers, is that they are the largest known kindred in the world with autosomal dominant Alzheimer’s disease, meaning it only takes one parent with the mutation to pass it onward. There are other, smaller cohorts as near as a neighboring town and as far away as Japan and Italy, each with their own “private” mutations.

“You could think of Alzheimer’s as basically one of two types,” Kosik said. “It’s more complex than that, but to start, there are families in which it’s clearly genetic — if you get the mutation, you get the disease. And there’s all the other cases, which we call sporadic.” There may be risks that are passed down, and lifestyle and aging can play big roles in the sporadic cases, but there is no completely penetrant genetic link, he explained.
“So the question is, is there a difference that we can detect between cases which are strongly genetic, and cases in which other factors may be involved?” Kosik said. “If you alter your genes in two different ways, one with a mutation from the time of conception and the others from your small risk and lifestyle, are they the same set of genes or are they different sets?”
Turns out, there are differences. Using state-of-the-art technology called single nucleus sequencing, which allows researchers to see which genes are turned on at the individual cell level, project scientist and lead author Camila Almeida sequenced the genes of brain cells with genetic Alzheimer’s. This allowed the researchers to compare those sequences with that of a control group with no Alzheimer’s, and with that from a group with sporadic Alzheimer’s. Meanwhile, co-lead author Sarah Eger did the statistical heavy lifting that enabled the researchers to contextualize the data.
An incomplete destruction
“There is a difference in that if you have a mutation causing Alzheimer’s, you have a preferential activation in many different cell types — neurons, astrocytes and other cells — that turn on an autophagy system involved in taking proteins that are bad, that are misfolded, that may be contributing to disease, and destroying them,” Kosik said. This, he explained, means that the body is somehow alerted to these faulty proteins and has initiated a system of protein destruction, which is a normal, protective function of the body, though the compensatory response is ultimately unsuccessful.
“The mutation is making a protein that’s not normal, so the cell turns on these other genes to destroy the mutant protein, but it doesn’t quite work,” Kosik explained. In sporadic cases, the same system is turned on, but to a lesser degree. “There’s something else that’s probably more complicated happening there that we don’t fully understand,” he said.

These findings imply that since the genetic processes and patterns involved in the autosomal dominant cases are rather distinct from those involved in sporadic cases, treatments and therapies in development for the genetic version of Alzheimer’s may not be effective for the sporadic cases, and vice-versa. This is important, as clinical trials for potential Alzheimer’s drugs have been tested on populations such as the Colombia kindred. “I would say we have to be careful about extrapolating clinical trial results from the
Colombian kindred because the disease mechanisms are a little different,” Kosik said.
A rare escapee
And then there’s Aliria Rosa Piedrahita de Villegas, a family member with the same mutation who defied the odds by living to her 70s without developing the dementia that cuts her relatives down before they reach 60. Much about how this remarkable woman managed to escape the disease despite having the PSEN1 E280A mutation is still a mystery, but thanks to her family’s donation of her brain to science, Kosik and his team are among several collaborations around the world uncovering clues as to how she accomplished that feat.
One clue that appeared when researchers examined her brain tissue: While Aliria had the same overproduction of senile plaques that the rest of her family had, the tangles of misfolded tau protein that typically accompany the plaques in the frontotemporal cortex of Alzheimer’s patients in her were relatively scant, keeping intact things like motor skills and executive function.
“She decoupled the two pathologies, and by having the plaques but not the tangles, she was spared dementia,” Kosik said. “It really pointed us to the fact that we better study the tangles; people can tolerate a lot of amyloid plaques the way she did, but once you get tangles, you’re in big trouble.”
Another promising line of inquiry lies in a second, equally rare mutation found in her cells called the Christchurch variant, named for the city in New Zealand where it was originally found. It’s a mutation to a gene that produces lipoproteins, called APOE (apolipoprotein E), which itself produces a protein that carries fats and cholesterol through the bloodstream.
“She had one gene that was turned on that no one else had in the rest of the people with the mutations, or even in the sporadic population,” Kosik said. They were shocked when they saw it.
“This gene is called LRP1,” Kosik said, adding that they were “puzzled and amazed” when they saw it because in the world of Alzheimer’s disease, LRP1 would more likely be a villain, encoding a receptor of the same name on the surface of the cells that binds to APOE but also takes up tau into cells. Previous research by the Kosik group revealed that suppressing LRP1 in mice models also reduced the chances that pathological tau would be taken up by neurons which would have then replicated the misfolded proteins and continued the process of neurodegeneration.
“But it was our thinking that was wrong, and not nature,” Kosik said. “Because it turns out that LRP1 was not increased in every cell type.” Indeed, they found an increase only in the astrocytes, star-shaped cells with neuroprotective functions. “What they probably do is destroy the tau,” Kosik explained. “So our hypothesis now is that the reason she was protected is because thanks to LRP1, her astrocytes could take up more tau and destroy it, and prevent it from spreading.”
The Kosik Lab is now working on proving this hypothesis.
Sonia Fernandez
Senior Science Writer
(805) 893-4765
sonia.fernandez@ucsb.edu
Share this article
Related Stories